作者:郑希元,李海霞
[摘要] 单克隆抗体技术在疾病诊断、药物开发中的应用是目前医药领域的一个研究热点,也为多种疾病的治疗提供了新的途径,目前已有多个单抗药物获FDA批准上市。本文以PD-1单抗药物为例,对该药物的专利布局进行分析,并对单抗药物在中国专利审查过程中的支持问题进行重点解读,以期为我国医药生物企业的专利布局和维护专利稳定性提供借鉴。
[关键词] 单克隆抗体;PD-1;单抗药物;支持问题;专利布局
一、单克隆抗体靶点 ~ PD-1受体
抗体作为介导体液免疫的重要效应分子,是B细胞接受抗原刺激后增殖分化为浆细胞所产生的糖蛋白。由于其在疾病的诊断、免疫防治及基础研究中的重要作用,人工制备抗体技术已被人们广泛关注。1975年,Kohler和Milstein建立了单克隆抗体(Monoclonal Antibody,m Ab)制备技术,使得大规模制备高特异性、均质性抗体成为可能。此技术是将产生特异性抗体的小鼠B细胞与骨髓瘤细胞融合,形成杂交瘤细胞,它既有骨髓瘤细胞大量扩增和永生的特性,又具备免疫B细胞合成和分泌特异性抗体的能力。由于来源于一个 B细胞,故经筛选克隆得到的杂交瘤细胞仅产生抗单一抗原表位的特异性抗体,称为单克隆抗体(以下简称单抗)。[1]
1986年,世界上首个单抗药物--用于治疗器官移植出现的排斥反应的抗cd3单抗OKT3获得美国FDA的上市批准,由此拉开了单抗药物发展的序幕。近年来,单抗药物在肿瘤、自身免疫病、心血管疾病等领域呈现出迅猛发展的态势,取得了巨大的成功,在高投入、高产出、高回报率的理念指引下,国内外各大制药公司均加大了投入力度。由于抗体药物具有特异性强、疗效显著及毒性低等特点,在临床中已被广泛应用。根据汤森路透最新数据,全球目前已获批及进入Ⅲ期临床研究的抗体类药物共有243个,其中121个品种已经获得美国FDA批准,占创新生物抗体药物的74.23%。如今,单抗已成为现代生物制药的重要组成部分,更是制药企业争相布局的"金矿产业"。预计2016年全球抗体类药物市场规模已超过1000亿美元。[2]
PD-1(细胞程序性死亡受体-1)受体是主要表达在活化T细胞上的免疫抑制性受体,与表达在肿瘤细胞表面的配体PD-L1结合,通过降低T细胞的免疫应答,使肿瘤免疫逃逸。[3]随着对PD-1/PD-L通路的深入研究,其在树突状细胞、T淋巴细胞、B淋巴细胞等中的作用逐渐被揭示,应用抗体阻断PD-1/PD-L信号通路对肿瘤及慢性病毒感染等疾病进行生物治疗已成为近年研究的热点。[4]以靶向PD-1及其配体PD-L1的单抗药物为代表的肿瘤免疫疗法被视为未来最有前途的肿瘤治疗方法之一,曾被美国《Science》杂志评为2013年全球十大科学突破性技术的榜首。肿瘤免疫疗法可以有效地克服现有的肿瘤靶向治疗药物(包括靶向类单抗)的耐药性问题。同时,其未来最大的潜力来自于与其他肿瘤疗法的联合使用,包括与化疗、放疗、靶向治疗药物、治疗性疫苗的联合使用。
二、PD-1单抗药物市场之争
PD-1受体由日本京都大学本庶佑(Honjo Tasuku)教授于1992年发现。2005年,日本小野制药和美国Medarex制药共同合作开发Nivolumab(商品名Opdivo®),并于2006年提交PCT申请,公开号为WO2006121168A1,之后陆续在日本、美国、中国以及欧洲等药品主流市场获得授权。2009年,百时美施贵宝斥资24亿美元收购Medarex制药,将Nivolumab项目收入囊中,其临床试验、知识产权与后续开发权利由百时美施贵宝所主导。2014年7月4日,Nivolumab获得PMDA(日本医药品医疗器械综合机构)的上市批准,同年12月22日获得FDA(美国食品药品监督管理局)的上市批准,2015年6月19日获得EMA(欧洲药品管理局)的上市批准,并由小野制药在日本销售,百时美施贵宝在美国和欧洲销售。该药批准的适应症为转移性黑色素瘤,非小细胞肺癌、晚期肾细胞癌、霍奇金淋巴瘤等。
此外,百时美施贵宝的竞争对手~默沙东于2009年收购先灵葆雅,获得MK-3475(即Pembrolizumab,商品名Keytruda®,一种新型的人源化Ig G4-κ型单克隆抗体,通过作用于PD-1,阻断PD-1/PD-L1通路,进而有助于人体免疫系统攻击肿瘤细胞)的后续开发权。该药于2014年9月4日获得FDA的上市批准,又于2015年7月17日获得EMA的上市批准,用于治疗转移性黑色素瘤、非小细胞肺癌、晚期黑色素瘤。[5]
然而,正当默沙东还沉浸在Keytruda®获得FDA批准的喜悦中时,百时美施贵宝一纸诉状将其告上美国联邦法庭:百时美施贵宝及其日本合作伙伴小野制药称,PD-1抑制剂的美国专利由小野制药获得,并已许可给美国百时美施贵宝公司,授权范围涵盖该药物在肿瘤领域的应用,默沙东侵犯了双方在日本上市的PD-1抑制剂Opdivo®的专利。据此,百时美施贵宝在起诉书中要求法院判决该产品侵权。对此,默沙东承认小野制药确实享有该产品的方法专利,但同时表示,该专利是无效的。经过几番厮杀,2017年1月,百时美施贵宝在专利纠纷中获胜,与默沙东达成和解协议,后者支付前者6.25亿美元及6.5%销售提成。百时美施贵宝赢下PD-1专利战,坐享Keytruda®数十亿美元销售分成。[6]
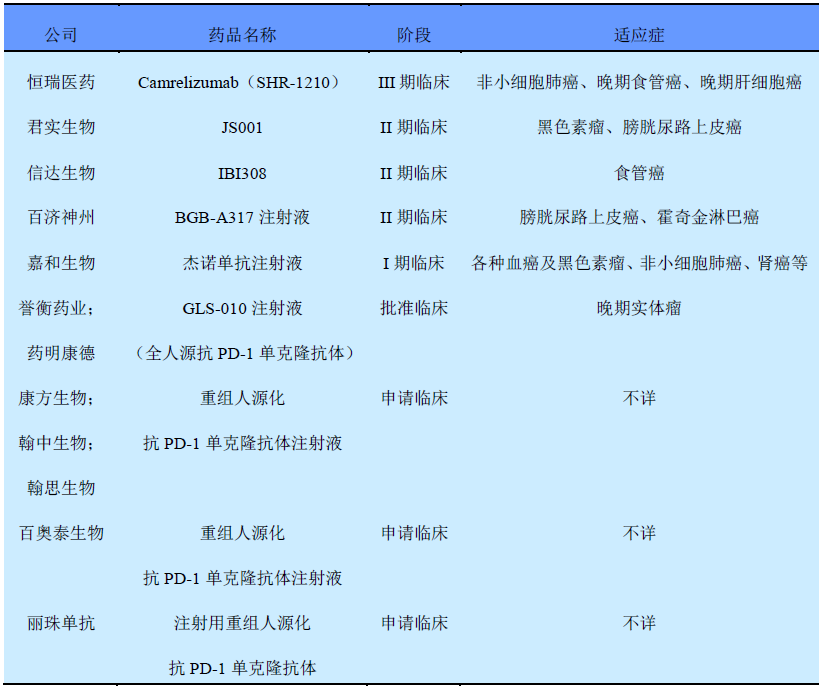
百时美施贵宝与默沙东在国际市场上战得难解难分,然后在中国市场,却是一片"和平"的景象。截至2017年4月21日,国内注册申报的PD-1单抗药物的制药公司共有9家。由于PD-1单抗药物是一类适应症很广的肿瘤免疫治疗药物,所以每家制药公司优先选择开发的适应症不尽相同(具体情况如表1所示,数据来源于"医药魔方数据")。
表1 国内已注册申报的PD-1抗体及其开发进度
究其原因,不妨来研究一下百时美施贵宝的日本合作伙伴小野制药在美国、欧洲和中国获得授权的同族专利的保护范围。
美国授权专利 ~ US8728474B2的权利要求1如下所示:
1. 一种用于治疗肿瘤患者的方法,包括给予患者药物有效量的抗-PD-1单克隆抗体。
欧洲授权专利 ~ EP1537878B1的权利要求1如下所示:
1. 抑制PD-1的免疫抑制信号的抗-PD-1抗体在制备用于癌症治疗的药物中的应用。
中国授权专利 ~ CN101213297B的权利要求1如下所示:
1. 人单克隆抗体或其抗原结合部分,
其包含
氨基酸序列如SEQ ID NO: 18所列的重链可变区CDR1;
氨基酸序列如SEQ ID NO: 25所列的重链可变区CDR2;
氨基酸序列如SEQ ID NO: 32所列的重链可变区CDR3;
氨基酸序列如SEQ ID NO: 39所列的轻链可变区CDR1;
氨基酸序列如SEQ ID NO: 46所列的轻链可变区CDR2;和
氨基酸序列如SEQ ID NO: 53所列的轻链可变区CDR3,
其中所述抗体或其抗原结合部分与人PD-1特异性结合。
比较上述权利要求的保护范围,不难发现小野制药欧洲授权专利的保护范围相比于中国授权专利的保护范围宽了不少,几乎是任何人未经允许将任何PD-1抗体用于任何癌症的治疗都将落入EP1537878B1的保护范围内并构成侵权,然而这样的权利要求在中国是无法获得授权的。
三、单抗药物中国专利审查中的"支持"问题
中国授权专利 ~ CN101213297B在发明专利申请公布时的权利要求1如下所示:
1. 分离的人单克隆抗体或其抗原结合部分,其中所述抗体与PD-1结合且其中所述抗体展现出至少一种下列性质:
a)以1×10-7 M或更小的KD与人PD-1结合;
b)不显著与CD28、CTLA-4或ICOS结合;
c)在混合淋巴细胞反应(MLR)测定法中增加T细胞增殖;
d)在MLR测定法中增加干扰素γ的产量;或
e)在MLR测定法中增加白介素-2(IL-2)的分泌。
由于该专利申请的年代较早,因此未能检索到该专利申请的具体审查档案,从而无法详细得知中国授权专利 ~ CN101213297B的权利要求1的具体修改和争辩过程。为了更清楚地了解单抗药物专利的中国审查过程,笔者另举一个中国专利申请 ~ CN102812047A的实例进行说明。
CN102812047A在发明专利申请公布时的权利要求1如下所示:
1. 分离的抗体或其抗原结合片段, 其与C4.4a的结构域S1特异性结合。
中国审查员在第一次审查意见通知书中指出:对比文件1(HANSEN L V ET AL, "Production, characterization and use of mono- and polyclonal antibodies against C4.4A, a homologue of the urokinase receptor", 10TH INTERNATIONAL WORKSHOP ON MOLECULAR AND CELLULAR BIOLOGY OF PLASMINOGEN ACTIVATION, 2005, 137)公开了与C4.4的结构域1(即本申请结构域S1)特异性结合的抗体。可见,权利要求1请求保护的技术方案已被对比文件1公开,且二者属于相同的技术领域,能够解决相同的技术问题,并达到相同的技术效果。因此权利要求1相对于对比文件1缺乏新颖性。
申请人在第一次审查意见答复过程中,将具备新颖性的权利要求2-4加入权利要求1中从而克服新颖性问题,修改后的权利要求1如下所示:
1. 分离的抗体或其抗原结合片段,其与C4.4a的结构域S1特异性结合,其中所述抗体或抗原结合片段与啮齿动物C4.4a交叉反应,其中所述抗体或其抗原结合片段在与表达C4.4a的细胞结合后被内化,其中所述抗体或抗原结合片段与抗体M31-B01或M20-D02 S-A竞争结合C4.4a,
a)其中所述抗体或其抗原结合片段包含与SEQ ID NO: 297 (CDR H1)、SEQ ID NO: 298 (CDR H2)、SEQ ID NO: 299 (CDR H3)一致的重链CDR序列及与SEQ ID NO: 300 (CDR L1)、SEQ ID NO: 22 (CDR L2)、SEQ ID NO: 301 (CDR L3)一致的轻链CDR序列,或者
b)其中所述抗体或其抗原结合片段包含与SEQ ID NO: 302 (CDR H1)、SEQ ID NO: 303 (CDR H2)、SEQ ID NO: 304 (CDR H3)一致的重链CDR序列及与SEQ ID NO: 305 (CDR L1)、SEQ ID NO: 306 (CDR L2)、SEQ ID NO: 307 (CDR L3)一致的轻链CDR序列。
中国审查员未接受上述修改,并在第二次审查意见通知书中指出:权利要求1采用了各CDR区的共有序列对所述抗体进行了限定。权利要求1限定的共有序列包括了多种序列的组合,其得到的含有六个CDR区的抗体的种类更是多样。然而包括六个CDR区的抗体的其中一个或多个CDR结构的改变都会导致抗体功能发生改变。本领域技术人员无法预期,除了说明书中所公开的抗体M31-B01(包含如SEQ ID NO: 5所示HCDR1,如SEQ ID NO: 9所示HCDR2,如SEQ ID NO: 13所示HCDR3,如SEQ ID NO: 17所示LCDR1、如SEQ ID NO: 21所示LCDR2,以及如SEQ ID NO: 25所示LCDR3)外,还有哪些抗体也能解决本发明的技术问题,并达到相同的技术效果。因此,权利要求1请求保护的技术方案得不到说明书的支持,不符合专利法第二十六条第四款的规定。
申请人在第二次审查意见答复过程中,修改了权利要求1,删除了CDR共有序列(SEQ ID NO:s 297-307),并将权利要求3的特征加入权利要求1中以克服支持问题,修改后的权利要求1如下所示:
1. 分离的抗体或其抗原结合片段,其与C4.4a的结构域S1特异性结合,其中所述抗体或抗原结合片段与啮齿动物C4.4a交叉反应,其中所述抗体或其抗原结合片段在与表达C4.4a的细胞结合后被内化,其中所述抗体或抗原结合片段与抗体M31-B01或M20-D02 S-A竞争结合C4.4a,
其中所述抗体或其抗原结合片段包含
SEQ ID NO: 75-77所示可变重链CDR序列和SEQ ID NO: 78-80所示可变轻链CDR序列,或者
SEQ ID NO: 5、9和13所示可变重链CDR序列和SEQ ID NO: 17、21和25所示可变轻链CDR序列,或者
......
SEQ ID NO: 45-47所示可变重链CDR序列和SEQ ID NO: 48-50所示可变轻链CDR序列,或者
......
SEQ ID NO: 135-137所示可变重链CDR序列和SEQ ID NO: 138-140所示可变轻链CDR序列。
中国审查员仍未接受上述修改,其在第三次审查意见通知书中除了指出"抗体的六个CDR区排列顺序应一一对应"之外,继续提出了与第二次审查意见通知书相同内容的权利要求1缺乏支持的审查意见。
申请人在第三次审查意见答复过程中,并没有选择审查员在审查意见中所指出的抗体M31-B01,而是选择将权利要求1修改为其想要保护的特定抗体B01-3,修改后的权利要求1如下所示:
1. 分离的抗体或其抗原结合片段,其与人C4.4a特异性结合,其中所述抗体或其抗原结合片段包含下列CDR序列:
SEQ ID NO: 45所示的CDR H1,
SEQ ID NO: 46所示的CDR H2,
SEQ ID NO: 47所示的CDR H3,
SEQ ID NO: 48所示的CDR L1,
SEQ ID NO: 49所示的CDR L2,和
SEQ ID NO: 50所示的CDR L3。
该专利申请的说明书第[0043]段和第[0403]段记载:图6提供了本发明抗体(B01-3)体外抑制肿瘤细胞增殖的数据。在实施例15描述的条件下,与未结合的hIgG1同种型对照抗体相比,抗体B01-3使细胞增殖明显降低。这就表明本发明的抗体有效抑制表达C4.4a的细胞的细胞增殖。
最终,审查员接受了申请人的修改,上述权利要求1最终获得了授权。
通过比较CN102812047A的最终授权的权利要求和CN101213297B的授权权利要求,不难发现,在中国的专利审查实践中,对于抗体产品的权利要求,通常要求用说明书中已经验证的特定抗体的重链和轻链可变区中的互补决定区序列来限定抗体。从CN102812047A的审查过程来看,这种对抗体权利要求的序列限定,通常是为了满足权利要求得到说明书支持的要求。尽管基于已有的限定特征已能够将请求保护的抗体区别于现有技术,中国审查员通常仍然会要求申请人将抗体限制为说明书中给出的特定的6个CDR序列。
从上面的实例可以看出,与美国和欧洲相对较为宽松的抗体专利授权标准(往往可以使用功能性限定)相比,中国对于抗体专利授权标准的要求相对较为严苛,即通常要求用抗体的重链和轻链的可变区中的三个高变区(即,互补性决定区,complementarity determining region,CDR)序列进行限定,属于序列限定型的权利要求。上述审查标准的确立也导致了只要中国企业研发的新PD-1抗体的CDR序列没有落入上述权利要求的范围,就基本上不存在侵权的问题。这也许就是PD-1抗体的市场争夺战在国际上激战正酣,而在中国市场上却风平浪静的重要原因。
四、PD-1抗体药物国内外专利布局比较分析
不仅美国和欧洲的抗体专利授权标准与中国不同,而且美国和欧洲的制药公司的抗体药物专利布局也与中国不尽相同。
仍以PD-1抗体药物为例,原研公司小野制药、Medarex和百时美施贵宝在布局氨基酸序列的核心专利时,均是通过PCT途径在全球主要市场同步进行申请,并尽量推迟专利公开的时间,以争取在产品上市前1~2年才让公众知晓其专利技术信息,以尽量减小被侵权的风险,同时还充分运用各国对药品专利的延长制度,最大限度延长专利保护时间,例如公开号为WO2006121168A1的氨基酸序列专利在美国获得授权后还获得了413天的延长期;在日本获得授权后还获得了接近5年的延长期,并通过对检测方法(例如WO2010001617A1,一种评价PD-1抗体治疗癌症效果的方法)、联合用药(例如WO2015134605A1,使用PD-1抗体和另一种抗癌剂治疗肾癌的方法;WO2016029073A2,PD-1抗体和CD137抗体联合治疗癌症的方法)、医药用途(例如WO2016100561A2,一种PD-1抗体治疗神经胶质瘤的方法)等外围专利的申请进行专利布局,编制庞大而且牢靠的专利保护网,提高技术门槛,为产品争取最大的保护强度。
除了原研公司之外,Incyte Corporation、葛兰素史克、Advaxis、辉瑞等公司主要通过申请联合用药专利进行布局(例如Incyte Corporation 的WO2015119944A1,一种治疗肿瘤的方法,包含PD-1拮抗剂和IDO1小分子药物;葛兰素史克的WO2015088847A1,一种用于治疗肿瘤的方法,包含PD-1拮抗剂和VFGFR抑制剂;Advaxis的WO2016011357A1,成细胞的灭活菌株治疗前列腺癌的方法;辉瑞的WO2016032927A1,一种治疗肿瘤的方法,包含PD-1拮抗剂和ALK 抑制剂(crizotinib)),其将PD-1抗体药物与本公司的核心产品通过复方制剂的方式进行二次研发,一定程度上绕开了原研公司的专利布局。但是这种改进型专利仍然存在一定的侵权风险,如果原研公司提出专利侵权诉讼,法院支持哪一方往往很难一概而论,其中的关键在于改进专利的那一方是否能够拿出足够的证据证明改进型专利在实施效果方面的显著优势,这样才能证明改进型专利优于原专利而没有侵犯原专利的权利。[5]
然而,表1中所提及的中国医药生物企业对于PD-1抗体的申请主要集中于国内申请,专利申请的技术分类也相对单一,主要集中在氨基酸序列专利和医药用途,具体情况如表2所示。
表2 国内企业对于PD-1抗体的申请状况

五、对我国医药生物企业的启示
单克隆抗体技术在疾病诊断、药物开发中的应用是目前医药领域的一个研究热点,也为多种疾病的治疗提供了新的途径,目前已有多个单抗药物获FDA批准上市,例如可用于头颈部肿瘤、结肠癌治疗的西妥昔单抗,可用于乳腺癌、胃食道癌治疗的曲妥珠单抗,可用于治疗慢性淋巴细胞性白血病的阿仑单抗。由于生物医药类产品科技含量高,附加值大,且普适性强,外国生物制药巨头一旦研发出新型单抗药物,就要进行专利保护,其保护范围往往涵盖全世界大多数国家,而中国作为最大的市场,更是专利保护的重中之重,故此,我国医药生物企业进行研发时已经面临严峻的专利垄断局面。
目前我国生产单抗药物的医药生物企业在专利战略上需要同时完成两个主要任务:既要防止外国生物制药巨头发起侵权诉讼,又要阻止国内的后来者轻易进行仿制。在实施上述专利战略的过程中,笔者认为可以从以下几个方面入手:
①我国医药生物企业应增强自主研发创新意识,积极开发针对新靶点或有效靶点新表位的单抗药物、积极研发并拓展现有单抗的适应症范围,并将自主研发的单抗药物通过PCT途径进行专利申请,并尽可能地推迟专利公布的时间,以争取在其产品上市前1~2年才让公众知晓其专利技术信息,从而尽可能地降低被侵权的风险,同时还可以充分运用各国对药品专利的延长制度,最大限度地延长专利保护时间;
②我国医药生物企业在开拓美国、欧洲等国外市场时,需时刻关注相关单抗药物专利的申请情况。由于外国生物制药巨头申请文件的权利要求中常常采用功能性限定、序列同源性限定、生物来源限定等各种方式,因此合理界定该单抗药物在世界各地的保护范围就显得尤为重要。在进行充分调研后再进行申请可以最大程度地确保专利权的稳定性;
③我国医药生物企业的知识产权部门应积极寻找自身已有专利的不足,通过对检测方法、联合用药、医药用途等外围专利的申请进行专利布局,建造牢固的专利保护体系,提高技术门槛,为自身产品争取最大的保护强度。同时可以利用"专利权无效宣告"途径来破解竞争对手的专利圈地;
④我国医药生物企业不仅应相互合作、联合研发,降低单抗药物的研发风险,而且应与国内各大高校、科研院所加强合作,进一步推进单抗药物的产学研相结合,加快研究成果的转化。
[参考文献]
[1] 陈慰峰, 金伯泉. 医学免疫学[M]. 人民卫生出版社, 2004.
[2] 金山. 抗体市场博弈好戏连台. 医药经济报[N]. 2017年1月9日(第006版).
[3] 魏木兰, 王艳林, 秦烨. PD-1/PD-L1抗体在肿瘤临床治疗中的应用. 生命科学[J]. 2016, 28(4): 475-479.
[4] 赵飞龙, 麦海星, 李学超, 陈立军, 张斌. PD-1/PD-L1信号通路在免疫细胞中的作用及其阻断抗体在肿瘤治疗中的应用. 细胞与分子免疫学杂志[J]. 2015, 31(5): 701-703.
[5] 从俊杰, 宿央央, 霍春芳, 刘沛然, 彭翠莲. PD-1/PD-L1抗体的专利分析. 今日药学[J]. 2017-04-28.
[6] 丁言. PD-1/PD-L1抗体烽烟再起. 医药经济报[N]. 2017年3月13日(第F02版).